Forschungsprojekte
General Research Topics
Methods of living/controlled ionic polymerization and ring-opening polymerization of heterocycles
Polymer modification, "thio-click" chemistry
Polymer and colloid characterization
Polymer colloids and films, composite materials
Bioinspired structure formation of polymers, hierarchical structures, polymer complexes
Polymer Synthesis:
Metal-free ring-opening polymerization of epoxides

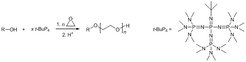
End-functionalized poly(ethylene oxide)s (PEG or PEO) and block copolymers are prepared by living/controlled anionic ring-opening polymerization of ethylene oxide using hydroxyl initiators in the presence of the phosphazene base t-BuP4 [Macromolecules 2001, 34, 4302; Langmuir 2003, 19, 4455; Adv. Mater. 2005, 17, 1158]. The t-BuP4 can also be used for a direct grafting of ethylene oxide from poly(N-isopropylacrylamide) (PNIPAM), using the NIPAM secondary amide units as initiating sites, to yield well-defined thermoresponsive graft copolymers [Macromolecules 2011, 44, 5861].
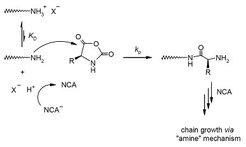
Ammonium-Mediated Polymerization of Amino Acid N-carboxyanhydrides
Block copolymers with a synthetic segment (polystyrene or 1,2-polybutadiene) and a polypeptide segment (poly(Z-L-lysine) or poly(γ-benzyl-L-glutamate)) are synthesized via the ring-opening polymerization of amino acid-N-carboxyanhydrides (NCA) initiated by primary amino-functional polymers. Using amine hydrochlorides instead of free amines yields block copolymers with near a monodisperse (Poisson) molecular weight distribution [Chem. Commun. 2003, 2944]. Kinetic studies show that the rate of polymerization is vastly determined by the position of the ammonium–amine equilibrium, thus it is higher the more polar is the reaction medium, the softer is the counterion, and the higher is the temperature [PMSE Prepr. 2007, 97, 183].

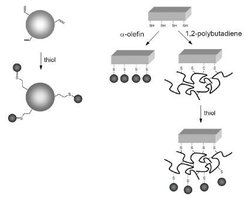
Modification of Unsaturated Polymers with Mercaptans, "Thio-Click" Polymer Chemistry
The modification of 1,2-polybutadienes through free-radical addition of mercaptans is a versatile strategy for the generation of a toolbox of functional polymers. However, the process suffers from a side reaction, namely formation of cyclic units along the backbone, affecting the degree of functionalization but not the molecular-weight distribution. Functional homopolymers and block copolymers carrying carboxylic acid, amine, ethylene glycol, perfluoroalkyl groups and also biohybrids with pendent amino acid, dipeptide, or glucose units have so been prepared [Polymer 2005, 46, 12057; Angew. Chem. Int. Ed. 2006, 45, 7578; J. Am. Chem. Soc. 2006, 128, 13336].
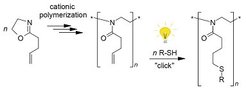
Replacing 1,2-polybutadiene by poly(2-[3-butenyl]-2-oxazoline), the photoaddition of mercaptans proceeds in the absence of side reactions and has the characteristics of a "click" reaction [Macromolecules 2007, 40, 7928; Polymer 2008, 49, 817].

The concept of radical thiol-ene photochemistry has been extended to the heterophase functionalization of polymer colloids and inorganic surfaces, i.e. glass fibers and slides. Particular emphasis was put on the preparation of sugar-containing materials [Chem. Eur. J. 2009, 15, 11469; Chem. Mater. 2009, 21, 5698].

Structure Formation:
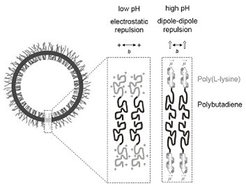
Polypeptide Block Copolymers
Polybutadiene-block-poly(L-lysine) can form large vesicles or "peptosomes" with a hydrodynamic radius of a few hundreds of nanometers in dilute aqueous saline solution. Vesicles are observed if the polypeptide segment is in a 100% coil conformation (pH 7.0) or in an 80% α-helical conformation (pH 10.3). At the higher pH, aggregates are smaller in size (hydrodynamic radius: 364 nm → 215 nm) and chains are more densely packed at the core-corona interface (inter-chain distance: 3.2 nm → 2.4 nm). Changes in size and structure can be explained in basic terms of colloid stabilization without considering a secondary structure effect [Langmuir 2007, 23 7196].

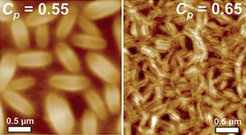
Swelling of thin films of polystyrene-poly(γ-benzyl-L-glutamate) heteroarm star block copolymers in chloroform vapour leads to the nucleation of ordered three-dimensional structures of ellipsoidal shape. The nucleation density not only increases with concentration but also sensitively depends on the presence of protic non-solvents, like for instance water, in the surrounding gas phase. An explanation for this behaviour is that the solubility of the polymer decreases upon hydrogen bonding complexation of water molecules to the polypeptide chains [Soft Matter 2008, 4, 993; collaboration with Günter Reiter].

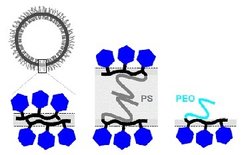
Amphiphilic glycopolymers
Glycopolymers, made by radical photoaddition of 1-thio-glucose onto 1,2-polybutadiene homopolymers and block copolymers, can self-assemble into vesicles or “glycosomes” in dilute aqueous solution, irrespective of their composition or hydrophilic weight fraction. Vesicles exhibit either a bilayered membrane or, in the case of glucosylated 1,2-polybutadiene-block-poly(ethylene oxide), a monolayered membrane with an asymmetric structure (i.e., glucose coating on the outside and PEO coating on the inside) [J. Am. Chem. Soc. 2006, 128, 13336; Macromolecules 2007, 40, 3901; Chem. Commun. 2009, 1478].

Hydrogen-Bonding Polymers
Glycosylated poly(2-oxazoline)s, prepared by combination of living cationic polymerization and "thio-click" chemistry, can self-assemble into spherical vesicles and nanofibers in dilute aqueous solution. The nanofibers are hollow nanotubes with a cross-sectional radius of about 3 nm and a wall with a thickness of about 1 nm. The wall should be constructed of chains forming a sheet through intermolecular hydrogen bonding between amide and glucose units [Macromol. Rapid Commun. 2008, 29, 304].


Poly[2-(acetoacetoxy)ethyl methacrylate] homopolymers, prepared by RAFT radical polymerization, can self-assemble into a hierarchical superstructure of double-stranded helical tubes with either screw sense. Both the diameter and pitch of the superhelices are about 12 nm and their length is in the order of 200-500 nm, depending on the molecular weight of the building polymer chain. Driven by hydrogen bridging interactions between adjacent acetoacetoxy groups and compensation of dipole moments, chains should first self-assemble into anti-parallel sheets or ribbons of ∼12 nm width and then coil up into the helical superstructure [J. Am. Chem. Soc. 2004, 26, 11307].

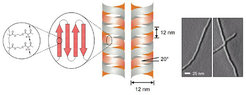
Crystallizing Polymers
Poly(2-isopropyl-2-oxazoline) is a thermoresponsive polymer exhibiting lower critical solution behavior in aqueous solution. However, long-term annealing of a dilute aqueous solution of poly(2-isopropyl-2-oxazoline) above its cloud point leads to the formation of coagulate in the form of crystalline nanofibers. The directional crystallization Process, which occurs below the glass transition of the polymer at 65 °C, is driven by hydrophobic and dipolar interactions in combination with a solvation effect [Soft Matter 2007, 3, 430; Angew. Chem. Int. Ed. 2007, 46, 8622; collaboration with Levent Demirel].


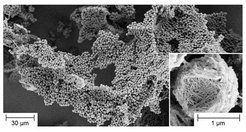
The crystallization of poly(2-isopropyl-2-oxazoline) in hot water can produce uniform microparticles with internal fibrous structure with a melting point close to 200 °C. The morphology on the micron length scale can be manipulated through the addition of a co-solvent or surfactant [Macromol. Rapid Commun. 2010, 31, 511].
Kinetic studies indicate that the temperature-induced phase separation of a dilute aqueous poly(2-isopropyl-2-oxazoline) solution produces a bicontinuous network-like structure, which with the onset of crystallization collapses into individual particles composed of a porous fiber mesh. These “premature” particles then act as nucleation sites for secondary crystallization. Nanofibers preferentially form at the particle surface, thus wrapping the microspheres like a ball of wool [Soft Matter 2010, 6, 3784].

Polyion Complexes
Mixing of polystyrene-block-poly(4-vinyl-N-methyl-pyridinium iodide) and polybutadiene-block-poly(cesium methacrylate) ionomers produces polyion complex vesicles in dilute tetrahydrofuran solution. Within the membrane of this novel species, polystyrene and polybutadiene segments are phase-separated, i.e. vesicles have a reduced symmetry and are amphiphilic in nature. Which of the segments is forming the solvating corona of the vesicles can be controlled by the selectivity of the solvent [Macromolecules 2003, 36, 1417].