Max-Planck-Institut für Kolloid- und Grenzflächenforschung
Biomimetische Nanostrukturen und -Prozesse: Computersimulation als bildgebendes Verfahren
Forschungsbericht (importiert) 2004 - Max-Planck-Institut für Kolloid- und Grenzflächenforschung
Autoren
Shillcock, Julian; Lipowsky, Reinhard
Abteilungen
Theorie und Bio-Systeme (Prof. Dr. Reinhard Lipowsky) MPI für Kolloid- und Grenzflächenforschung, Potsdam
Zusammenfassung
Biomimetische Systeme mit maßgeschneiderten Eigenschaften haben eine Vielzahl von potenziellen Anwendungen, etwa als Wirkstoffträger oder als Biosensoren. Die Konstruktion derartiger Systeme erfordert ein genaues Verständnis der zu Grunde liegenden Nanostrukturen und -prozesse. Diese Strukturen sind oft nur einige Nanometer dick und deshalb sehr flexibel und beweglich. Da es keine experimentelle Methode gibt, mit der man die Dynamik derartiger Nanostrukturen direkt abbilden könnte, bieten sich Computersimulationen als bildgebende Verfahren an. Eine besonders leistungsfähige Simulationsmethode ist die Dissipative-Partikel-Dynamik, die man einsetzen kann, um supramolekulare Systeme mit einer Lineardimension von 50 Nanometern über einen Zeitraum von mehreren Mikrosekunden zu beobachten. In diesem Beitrag werden drei Beispiele für derartige Systeme diskutiert: Lipidmembranen, die mehrere Komponenten enthalten und verschiedene Domänen ausbilden; Vesikel aus Diblock-Copolymeren, die als Wirkstoffträger interessant sind; sowie die Spannungs-induzierte Fusion von Doppelschicht-Membranen. Die Simulations-Methode der Dissipativen-Partikel-Dynamik kann zur Optimierung von Nanostrukturen und -prozessen eingesetzt werden, bevor man kostspielige experimentelle Versuchsreihen durchführt.
Einleitung
Die wissenschaftliche Untersuchung von biomimetischen Systemen verfolgt zwei komplementäre Ziele: ein tieferes Verständnis der belebten Natur sowie eine Verbesserung der menschlichen Lebensbedingungen. Für beide Ziele spielt die Visualisierung von Strukturen und Prozessen im Nanobereich eine wichtige Rolle. Auf der einen Seite beschäftigen sich die Biowissenschaften mit komplexen Molekülverbänden in lebenden Zellen. Diese supramolekularen Strukturen haben ungewöhnliche Architekturen, die sich ohne Visualisierung nicht verstehen lassen. Auf der anderen Seite gibt es viele Gebrauchsgüter der chemischen, kosmetischen und pharmazeutischen Industrie, die aus biomimetischen Nanostrukturen aufgebaut sind. Um die Eigenschaften dieser Erzeugnisse zu optimieren, möchte man „sehen“ können, wie sie auf der Nanoskala mit biologischen Zellen wechselwirken.
Leider ist es nicht möglich, das Verhalten dieser Systeme direkt im Lichtmikroskop zu beobachten, da die Strukturen deutlich dünner sind als die Wellenlängen von sichtbarem Licht. Die Elektronenmikroskopie ermöglicht es im Prinzip, 1000-mal kleinere Strukturen aufzulösen. Allerdings kann sie nur statische Schnappschüsse liefern und ist deshalb nicht geeignet, die vielen dynamischen Prozesse im Nanobereich zu verfolgen. Die gleiche Einschränkung gilt für die verschiedenen Kraftmikroskopie-Methoden mit ultrafeinen Abtastnadeln, die nur Strukturen abbilden können, die zuvor auf einem festen Substrat immobilisiert worden sind. Hier bietet sich die Computersimulation als wichtiges Werkzeug an, um sowohl die Struktur als auch die Dynamik von Bionanosystemen sichtbar zu machen [1].
Ein wichtiger Beitrag zum Verständnis solcher Systeme wurde von Computeralgorithmen der Molekular-Dynamik mit atomarer Auflösung (all atom simulations) geleistet. Bei dieser Simulationsmethode werden alle Atome, aus denen ein bestimmtes System aufgebaut ist, berücksichtigt. Jedes Atom hat eine bestimmte Masse, Größe, Position und Geschwindigkeit. Die Atome üben Kräfte aufeinander aus und bewegen sich gemäß der Newton’schen Gesetze. In der Computersimulation wird das Verhalten von Tausenden von Atomen berechnet. Gleichzeitig kann man die kooperativen Eigenschaften dieser Systeme, also z.B. ihre mechanischen oder rheologischen Eigenschaften, in der Simulation verfolgen und mit geeigneten Mittelungsverfahren „messen“. Die Simulationsmethode der Molekular-Dynamik ist jedoch sehr kostspielig, da die benötigte Rechenzeit sehr schnell mit der Systemgröße ansteigt. So benötigen Molekular-Dynamik-Simulationen, die die Bewegung von einigen hundert Seifenmolekülen in Wasser für einige Nanosekunden verfolgen, mehrere tausend Stunden Rechenzeit.
In den letzten zehn Jahren wurden allerdings neue Simulationstechniken entwickelt, die die Beschränkungen, denen die molekulare Dynamik unterliegt, überwinden. Eine dieser Methoden, die so genannte Dissipative-Partikel-Dynamik, wurde 1992 [2] erfunden und fand breite Anwendung bei der Simulation biomimetischer Systeme. So können ganz unterschiedliche Systeme wie Polymere in Lösung [3] oder Lipidmembranen im Wasser [4] simuliert und visualisiert werden. Die Dissipative-Partikel-Dynamik erlaubt es, Millionen von Molekülen mit Volumina von 1 nm3 bis 106 nm3 über einige Mikrosekunden hinweg zu beobachten. Dabei wird nur ein Bruchteil der Computerressourcen eingesetzt, die für die konventionelle Molekular-Dynamik dieser Systeme benötigt würden.
Von Seifenfilmen und Lipidmembranen
Seifen und Lipide sind Beispiele für Amphiphile, d.h. für Moleküle die zwei verschiedene Enden besitzen. Das eine Ende ist hydrophil und „liebt“ das Wasser, das andere Ende ist hydrophob und vermeidet den Kontakt mit demselben. Da die beiden Enden miteinander verbunden sind, ordnen sich die Moleküle im Wasser so an, dass beide Enden möglichst „zufrieden“ sind. Deshalb heften sich Seifenmoleküle an ölige Schmutzpartikel so an, dass die hydrophilen Enden zum Wasser zeigen. Auf diese Weise werden diese Partikel wasserlöslich und können von der Haut abgelöst werden. Lipidmoleküle im Wasser bilden typischerweise Doppelschicht-Membranen, siehe Abbildung 1, bei denen sich die hydrophilen Enden gegenüber liegen. Diese Doppelschichten, die nur eine Dicke von etwa 4 Nanometern haben, bilden spontan geschlossene Blasen oder Vesikel aus. Auch biologische Zellen und Zellorganellen sind von derartigen Doppelschicht-Membranen umgeben, die als flexible aber dennoch robuste Barrieren fungieren. Des Weiteren bilden die Membranen ein flüssiges Medium, in dem die Membranproteine ungehindert ihre Wirkung entfalten können.
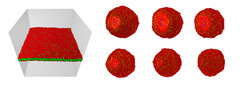
(Links) Orientiertes Membransegment, das aus etwa 8000 Lipidmolekülen besteht und von etwa 3 Millionen Wassermolekülen umgeben ist (das Wasser ist nicht abgebildet). Das Segment durchspannt die Simulationsbox, von der vier Seitenwände zu sehen sind. Die laterale Ausdehnung des Segments beträgt 50 Nanometer. Die Lipidmoleküle bilden eine Doppelschicht-Struktur aus, bei der die hydrophoben Ketten (grüne Teilchen) zur Membranmitte hin zeigen. (Rechts) Sechs Schnappschüsse einer Membranblase oder Vesikel mit einem Durchmesser von etwa 28 Nanometern; die Membran enthält etwa 7400 Lipidmoleküle. Die Sequenz beginnt links oben und endet rechts unten. Am Anfang hat die Vesikelmembran eine Pore (hellrot), die sich zunächst weiter öffnet und danach wieder schließt. Derartige „Selbstheilungsprozesse“ werden auch experimentell beobachtet. Alle hier gezeigten Membranen sind fluide: benachbarte Lipidmoleküle tauschen ihre Plätze innerhalb von 100 Nanosekunden.
(Links) Orientiertes Membransegment, das aus etwa 8000 Lipidmolekülen besteht und von etwa 3 Millionen Wassermolekülen umgeben ist (das Wasser ist nicht abgebildet). Das Segment durchspannt die Simulationsbox, von der vier Seitenwände zu sehen sind. Die laterale Ausdehnung des Segments beträgt 50 Nanometer. Die Lipidmoleküle bilden eine Doppelschicht-Struktur aus, bei der die hydrophoben Ketten (grüne Teilchen) zur Membranmitte hin zeigen. (Rechts) Sechs Schnappschüsse einer Membranblase oder Vesikel mit einem Durchmesser von etwa 28 Nanometern; die Membran enthält etwa 7400 Lipidmoleküle. Die Sequenz beginnt links oben und endet rechts unten. Am Anfang hat die Vesikelmembran eine Pore (hellrot), die sich zunächst weiter öffnet und danach wieder schließt. Derartige „Selbstheilungsprozesse“ werden auch experimentell beobachtet. Alle hier gezeigten Membranen sind fluide: benachbarte Lipidmoleküle tauschen ihre Plätze innerhalb von 100 Nanosekunden.
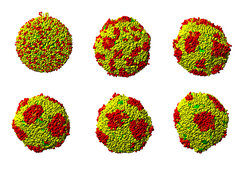
Vor kurzem konnten Simulationen der Dissipativen-Partikel-Dynamik eingesetzt werden, um die Materialeigenschaften von Doppelschicht-Membranen zu untersuchen, die mehrere tausend Lipidmoleküle enthalten [4]. Dabei wurde der Einfluss der molekularen Lipidstruktur auf die Membransteifigkeit und -dehnbarkeit besonders deutlich. Ausgehend von diesen ersten Versuchen konnten Membranen, die sich aus zwei verschiedenen Lipiden zusammensetzen, simuliert werden. Abbildung 2 illustriert das Wachstum von Domänen in einer quasi-sphärischen Vesikel, deren Membran zwei unterschiedliche Lipidmoleküle enthält [5, 6]. Die beiden Molekülsorten sind in den roten und gelben Domänen angereichert. Die elastischen Eigenschaften von zweikomponentigen Membranen wurden auch mit Molekular-Dynamik-Simulationen untersucht, wobei hier ebenfalls mehrere Atome zu Atomgruppen zusammengefasst wurden [7].
Domänenwachstum in einer Vesikelmembran mit zwei Lipidkomponenten, deren Kopfgruppen rot bzw. gelb dargestellt sind. Die Sequenz aus sechs Schnappschüssen beginnt links oben mit einer zufälligen Anordnung der Lipidmoleküle in der Membran und endet rechts unten mit klar abgegrenzten Domänen der „roten“ Lipide. Der hier abgebildete Prozess dauert einige Mikrosekunden [6].
Domänenwachstum in einer Vesikelmembran mit zwei Lipidkomponenten, deren Kopfgruppen rot bzw. gelb dargestellt sind. Die Sequenz aus sechs Schnappschüssen beginnt links oben mit einer zufälligen Anordnung der Lipidmoleküle in der Membran und endet rechts unten mit klar abgegrenzten Domänen der „roten“ Lipide. Der hier abgebildete Prozess dauert einige Mikrosekunden [6].
Lipidvesikeln werden schon seit längerem als potenzielle Transportsysteme für verschiedene Wirkstoffe untersucht, besitzen aber zwei Nachteile. Erstens kann die Lipidmembran nur um einige wenige Prozent gedehnt werden, ohne dass sie aufreißt. Daraus ergibt sich eine besondere Anfälligkeit der Vesikel bei Änderungen des osmotischen Drucks. Zweitens haben membranbildende Lipide eine bestimmte molekulare Architektur, die nur in gewissen Grenzen variiert werden kann. Vesikel aus Diblock-Copolymeren (so genannte Polymersome) bieten hier einen alternativen Zugang. Das Molekulargewicht und das hydrophobe/hydrophile Blockverhältnis kann dabei über einen weiten Bereich variiert werden. Diese Eigenschaften machen Polymersome sehr interessant für die Verkapselung von Wirkstoffen. Allerdings fehlt es nach wie vor an Detailwissen über den Zusammenhang von Polymerarchitektur und Vesikel-Eigenschaften.
Zur Aufklärung dieses Zusammenhangs wurden wieder Simulationen der Dissipativen-Partikel-Dynamik eingesetzt [8]. Die Parameter der Dissipativen-Partikel-Dynamik lassen sich aus denen der Molekular-Dynamik mithilfe einer neuen Methode ableiten. Dabei wird die Dichte der verschiedenen Partikelarten so angepasst, dass die in Experimenten gemessenen Moleküldichten in der Doppelschicht reproduziert werden. Benutzt man die neu abgeleiteten Parameter, kann man verschiedene Materialeigenschaften von Polymersomen mithilfe der Simulationen untersuchen, so etwa das Zerreißen der Vesikel-Membran, wenn die Vesikel mit Wasser aufgebläht wird. Abbildung 3 zeigt eine Polymersom-Vesikel, die nahezu 8000 Diblock-Copolymere enthält. Wenn diese Vesikel am Anfang zu viel Wasser enthält, dann bildet sich eine Membran-Pore, aus der das überschüssige Wasser austritt, siehe Abbildung 3. Es ist relativ einfach, in den Simulationen die Polymerarchitektur zu variieren. Auf diese Weise lassen sich Polymersome für den Einsatz als Wirkstoffträger optimieren, bevor man kostspielige experimentelle Versuchsreihen durchführt.
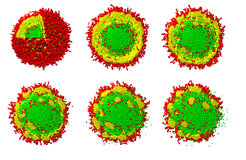
(Links oben) Vesikel mit einer Membran aus 2018 Diblock-Copolymeren; die Vesikel hat einen Durchmesser von 40 nm. Das hier verwendete Polymer heißt PEO40PEE37 und verknüpft 40 PEO-Monomere (rote Ketten), die hydrophil sind, mit 37 PEE-Monomeren (gelbe Ketten), die hydrophob sind. Das Wasser im Innern der Vesikel ist grün eingefärbt, das Wasser im äußeren Kompartiment ist nicht abgebildet. Die weiteren fünf Schnappschüsse zeigen die zeitliche Entwicklung der Membranstruktur, wenn die Vesikel am Anfang zu viel Wasser enthält und deshalb stark gespannt ist. Diese Spannung führt zur Poration der Membran; im letzten Schnappschuss (rechts unten) sind einige „grüne“ Wassermoleküle zu erkennen, die durch die Pore ausgetreten sind [8].
(Links oben) Vesikel mit einer Membran aus 2018 Diblock-Copolymeren; die Vesikel hat einen Durchmesser von 40 nm. Das hier verwendete Polymer heißt PEO40PEE37 und verknüpft 40 PEO-Monomere (rote Ketten), die hydrophil sind, mit 37 PEE-Monomeren (gelbe Ketten), die hydrophob sind. Das Wasser im Innern der Vesikel ist grün eingefärbt, das Wasser im äußeren Kompartiment ist nicht abgebildet. Die weiteren fünf Schnappschüsse zeigen die zeitliche Entwicklung der Membranstruktur, wenn die Vesikel am Anfang zu viel Wasser enthält und deshalb stark gespannt ist. Diese Spannung führt zur Poration der Membran; im letzten Schnappschuss (rechts unten) sind einige „grüne“ Wassermoleküle zu erkennen, die durch die Pore ausgetreten sind [8].
Einblick in zelluläre Membranprozesse: Vesikelfusion
An der Vesikelfusion in vivo sind verschiedene Proteine beteiligt, die auf komplexe Weise miteinander wechselwirken. Allerdings können auch einfache Lipidmembranen miteinander fusionieren, wenn sie unter Spannung stehen. In beiden Fällen beginnt die Membranfusion mit der Bildung einer Fusionspore, einem Ereignis, das sich auf einer Längenskala von 10 bis 20 Nanometern und auf einer Zeitskala, die unter einer Millisekunde liegt, abspielt. Es ist deshalb bislang nicht möglich, die Bildung von Fusionsporen direkt experimentell zu beobachten.
Mithilfe der Dissipativen-Partikel-Dynamik ist es nun gelungen, den zeitlichen Verlauf der Membranfusion auf der Nanometer-Skala systematisch zu untersuchen [9]. Dabei wurden die Spannungen in den beiden Doppelschicht-Membranen als Kontrollparameter verwendet. Abbildung 4 zeigt ein typisches Fusionsereignis zwischen einer gespannten Vesikel und einem ebenfalls gespannten Membransegment. Insgesamt wurden 93 derartige Fusionsversuche simuliert und ausgewertet. Die Ergebnisse dieser Simulationen lassen sich in einem globalen Morphologie-Diagramm zusammenfassen, siehe Abbildung 5. Aus diesem Diagramm kann man ablesen, wie die Fusionswahrscheinlichkeit von den Spannungen in den beiden Membranen abhängt.
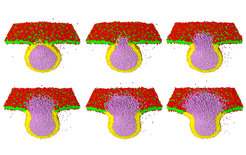
Sequenz von sechs Schnappschüssen, aufgenommen während einer typischen Fusion von einer Vesikel (Durchmesser 28 nm) und einem quadratischen Membransegment (lineare Ausdehnung 50 nm). Die beiden Membranen bestehen aus 5684 bzw. 5455 Lipiden und werden zu Beginn der Simulation einer Spannung ausgesetzt, die die molekulare Fläche vergrößert. In diesem Beispiel beträgt die anfängliche, dimensionslose Fläche pro Molekül A/Na02 = 1,9 und 1,45 für das Membransegment bzw. für die Vesikel. Dabei ist A die Membranfläche, N die Anzahl der Moleküle und a0 die Größe der simulierten Teilchen. Die Wassermoleküle außerhalb der Vesikel sind transparent und unsichtbar, während die Wassermoleküle im Inneren der Vesikel als violette Teilchen zu sehen sind. Der erste Schnappschuss wurde 80 Nanosekunden nach Simulationsbeginn aufgenommen, die folgenden Schnappschüsse nach jeweils 32 weiteren Nanosekunden.
Sequenz von sechs Schnappschüssen, aufgenommen während einer typischen Fusion von einer Vesikel (Durchmesser 28 nm) und einem quadratischen Membransegment (lineare Ausdehnung 50 nm). Die beiden Membranen bestehen aus 5684 bzw. 5455 Lipiden und werden zu Beginn der Simulation einer Spannung ausgesetzt, die die molekulare Fläche vergrößert. In diesem Beispiel beträgt die anfängliche, dimensionslose Fläche pro Molekül A/Na02 = 1,9 und 1,45 für das Membransegment bzw. für die Vesikel. Dabei ist A die Membranfläche, N die Anzahl der Moleküle und a0 die Größe der simulierten Teilchen. Die Wassermoleküle außerhalb der Vesikel sind transparent und unsichtbar, während die Wassermoleküle im Inneren der Vesikel als violette Teilchen zu sehen sind. Der erste Schnappschuss wurde 80 Nanosekunden nach Simulationsbeginn aufgenommen, die folgenden Schnappschüsse nach jeweils 32 weiteren Nanosekunden.
Das Verhalten, das in diesen Fusionssimulationen beobachtet wurde, war in mehrfacher Hinsicht überraschend. Einerseits gibt es für die untersuchte Membrangeometrie nur einen relativ kleinen Spannungsbereich, in dem Fusion auftreten kann, siehe Abbildung 5. Aber selbst in diesem Bereich läuft der Fusionsprozess stochastisch ab: etwa 50 Prozent der Fusionsversuche führten in diesem Bereich nicht zur Fusion, sondern zum Zerreißen der Vesikel, zum Zerreißen des Membransegments, oder zur Hemifusion der beiden Doppelschichten. Außerdem stellte sich heraus, dass alle erfolgreichen Fusionsversuche innerhalb von 200-300 Nanosekunden abliefen. So konnten keine Fusionen mit einer Fusionszeit oberhalb von 350 Nanosekunden beobachtet werden. Diese obere Schranke für die Fusionszeit hängt mit der Stabilisierung des hemifusionierten Zustandes durch die betrachtete Membrangeometrie zusammen und sollte auch für die Fusion von zwei Vesikeln gelten, die eine vergleichbare Größe haben [9].
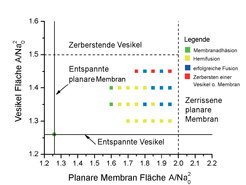
Morphologiediagramm für eine Vesikel im Kontakt mit einer planaren Membran als Funktion der anfänglichen Membranfläche pro Molekül A/Na02 für die planare Membran und die Vesikel. Diese Molekülflächen bestimmen die Spannungen in den Membranen. Die entspannten Zustände von Vesikel und Membransegment entsprechen den beiden geraden Linien, die sich in dem grünen Punkt links unten schneiden. Die beiden gestrichelten Linien entsprechen dem Zerreißen des planaren Membransegments bzw. der Vesikel. Die Farbcodierung der Punkte im Diagramm zeigt an, welcher Prozess bei den vorgegebenen Molekülflächen dominiert: erfolgreiche Fusion (blaue Quadrate); Zerbersten einer Vesikel oder einer planaren Membran (rote Quadrate); Hemifusion (gelbe Quadrate) und Membranadhäsion (grüne Quadrate). Insgesamt wurden 93 unabhängige Simulationen durchgeführt, um die im Diagramm gezeigten Daten zu generieren [9].
Morphologiediagramm für eine Vesikel im Kontakt mit einer planaren Membran als Funktion der anfänglichen Membranfläche pro Molekül A/Na02 für die planare Membran und die Vesikel. Diese Molekülflächen bestimmen die Spannungen in den Membranen. Die entspannten Zustände von Vesikel und Membransegment entsprechen den beiden geraden Linien, die sich in dem grünen Punkt links unten schneiden. Die beiden gestrichelten Linien entsprechen dem Zerreißen des planaren Membransegments bzw. der Vesikel. Die Farbcodierung der Punkte im Diagramm zeigt an, welcher Prozess bei den vorgegebenen Molekülflächen dominiert: erfolgreiche Fusion (blaue Quadrate); Zerbersten einer Vesikel oder einer planaren Membran (rote Quadrate); Hemifusion (gelbe Quadrate) und Membranadhäsion (grüne Quadrate). Insgesamt wurden 93 unabhängige Simulationen durchgeführt, um die im Diagramm gezeigten Daten zu generieren [9].
Die Simulationen machen deutlich, dass die von relativ großen Spannungen induzierte Fusion sehr schnell abläuft. Allerdings sind diese Fusionsprozesse stochastisch und unzuverlässig. Ein verlässlicheres Fusionsprotokoll scheint fein abgestimmte Kräfte zu erfordern, die örtlich beschränkt und nur über ein relativ kurzes Zeitintervall wirksam sind. Derartige Kräfte sollten durch die Proteine, die die Fusion in vivo induzieren, ausgeübt werden. Hier werden Simulationen für die Fusion von mehrkomponentigen Membranen zusätzliche Einsichten bringen.
Die obigen Beispiele zeigen, dass Partikel-basierte Simulationsmethoden in der Lage sind, biomimetische Strukturen und Prozesse im Nanobereich sichtbar zu machen. Diese Methoden werden ständig verfeinert und weiterentwickelt und werden sicher noch viele neue Einblicke in diesen Bereich ermöglichen. Dieses Verständnis bildet eine wichtige Voraussetzung für das rationale Design von Wirkstoffmolekülen und -trägern, künstlichen Membranen und anderen biomimetischen Nanosystemen.
Originalveröffentlichungen
1.
Lipowsky, R.
Biomimetic membrane modelling: Pictures from the twilight zone
Nature Materials 3, 589-591 (2004).
2.
Hoogerbrugge, P. J.; Koelman, J. M. V. A.
Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics
Europhysics Letters 19, 155-160 (1992).
3.
Groot, R. D.; Warren, P. B.
Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation
Journal of Chemical Physics 107, 4423-4435 (1997).
4.
Shillcock, J. C.; Lipowsky, R.
Equilibrium structure and lateral stress distribution of amphiphilic bilayers from dissipative particle dynamics simulations
Journal of Chemical Physics 117, 5048-5061 (2002).
5.
Illya, G.; Lipowsky, R.; Shillcock, J. C.
Effect of chain length and asymmetry on bilayer material properties
Journal of Chemical Physics, to appear in Vol. 122 (2005).
6.
Illya, G.
Bilayer material properties and domain formation from dissipative particle dynamics
Dissertation, Universität Potsdam (2004).
7.
Imparato, A.; Shillcock, J.; Lipowsky, R.
Shape fluctuations and elastic properties of two-component bilayer membranes
Europhysics Letters 69, 650-656 (2005).
8.
Ortiz, V.; Nielsen, S. O.; Discher, D. E.; Klein, M. L.; Lipowsky, R.; Shillcock; J. C.
Dissipative particle dynamics simulations of polymersomes: Structural and mechanical properties
Journal of Physical Chemistry B (submitted, 2005).
9.
Shillcock, J. C.; Lipowsky, R.
Tension-induced fusion of bilayer membranes and vesicles
Nature Materials 4, 225-228 (2005).
10.
Die Schnappschüsse in den Abbildungen wurden mit dem PovRay Suchprogramm erstellt.
 Orientiertes Membransegment, das aus etwa 8000 Lipidmolekülen besteht und von etwa 3 Millionen Wassermolekülen umgeben ist (das Wasser ist nicht abgebildet). Das Segment durchspannt die Simulationsbox, von der vier Seitenwände zu sehen sind. Die laterale Ausdehnung des Segments beträgt 50 Nanometer. Die Lipidmoleküle bilden eine Doppelschicht-Struktur aus, bei der die hydrophoben Ketten (grüne Teilchen) zur Membranmitte hin zeigen. (Rechts) Sechs Schnappschüsse einer Membranblase oder Vesikel mit einem Durchmesser von etwa 28 Nanometern; die Membran enthält etwa 7400 Lipidmoleküle. Die Sequenz beginnt links oben und endet rechts unten. Am Anfang hat die Vesikelmembran eine Pore (hellrot), die sich zunächst weiter öffnet und danach wieder schließt. Derartige „Selbstheilungsprozesse“ werden auch experimentell beobachtet. Alle hier gezeigten Membranen sind fluide: benachbarte Lipidmoleküle tauschen ihre Plätze innerhalb von 100 Nanosekunden.")

 Vesikel mit einer Membran aus 2018 Diblock-Copolymeren; die Vesikel hat einen Durchmesser von 40 nm. Das hier verwendete Polymer heißt PEO40PEE37 und verknüpft 40 PEO-Monomere (rote Ketten), die hydrophil sind, mit 37 PEE-Monomeren (gelbe Ketten), die hydrophob sind. Das Wasser im Innern der Vesikel ist grün eingefärbt, das Wasser im äußeren Kompartiment ist nicht abgebildet. Die weiteren fünf Schnappschüsse zeigen die zeitliche Entwicklung der Membranstruktur, wenn die Vesikel am Anfang zu viel Wasser enthält und deshalb stark gespannt ist. Diese Spannung führt zur Poration der Membran; im letzten Schnappschuss (rechts unten) sind einige „grüne“ Wassermoleküle zu erkennen, die durch die Pore ausgetreten sind [8].")
 und einem quadratischen Membransegment (lineare Ausdehnung 50 nm). Die beiden Membranen bestehen aus 5684 bzw. 5455 Lipiden und werden zu Beginn der Simulation einer Spannung ausgesetzt, die die molekulare Fläche vergrößert. In diesem Beispiel beträgt die anfängliche, dimensionslose Fläche pro Molekül A/Na02 = 1,9 und 1,45 für das Membransegment bzw. für die Vesikel. Dabei ist A die Membranfläche, N die Anzahl der Moleküle und a0 die Größe der simulierten Teilchen. Die Wassermoleküle außerhalb der Vesikel sind transparent und unsichtbar, während die Wassermoleküle im Inneren der Vesikel als violette Teilchen zu sehen sind. Der erste Schnappschuss wurde 80 Nanosekunden nach Simulationsbeginn aufgenommen, die folgenden Schnappschüsse nach jeweils 32 weiteren Nanosekunden.")
; Zerbersten einer Vesikel oder einer planaren Membran (rote Quadrate); Hemifusion (gelbe Quadrate) und Membranadhäsion (grüne Quadrate). Insgesamt wurden 93 unabhängige Simulationen durchgeführt, um die im Diagramm gezeigten Daten zu generieren [9].")