Mehrskalen-Modellierung von Polysacchariden und Polysaccharidmaterialien
Forschungsbericht (importiert) 2014 - Max-Planck-Institut für Kolloid- und Grenzflächenforschung
Biomoleküle stellen eine große Herausforderung für die Modellierung dar, da sie unterschiedliche inhärente Skalen besitzen. Kleinste Veränderungen der Molekülbausteine haben große Auswirkungen auf die Materialeigenschaften. So zeigen z. B. natürliche Polysaccharide, die aus wenigen einfachen Zuckerbausteinen bestehen, außergewöhnliche Materialeigenschaften. Diese hängen von der räumlichen Anordnung der Zuckerbausteine ab. Geeignete Modelle müssen eine atomistische Beschreibung mit vergröberten Darstellungen kombinieren, die möglichst viele Charakteristika des hochaufgelösten Systems erhalten.
Polysaccharide: Polymere mit vielseitigem Einsatz
Polysaccharide oder Vielfachzucker gehören zu den in der Natur am häufigsten vorkommenden Polymeren. Ihre Funktion in lebenden Organismen ist dabei erstaunlich vielfältig [1]. Am bekanntesten ist ihre Rolle als Energielieferant und Energiespeicher, aber Polysaccharide erfüllen weit vielfältigere Aufgaben. So ist z. B. jede Zelle von einer sogenannten Glycokalix umgeben. Dies ist eine etwa 100 nm dicke Kohlenhydratschicht, die sowohl dem Schutz der Zelle dient als auch an der Zell-Zell-Kommunikation beteiligt ist. Proteoglykane, mit vielen Polysacchariden versehene Proteine, bilden einen Hauptbestandteil der extrazellulären Matrix und der erste Kontakt mit Pathogenen involviert vor allem Zuckermoleküle. In Pflanzen und Insekten übernehmen Polysaccharidmaterialien wie Zellulose und Chitin außerdem wichtige strukturelle Funktionen.


Komplexe Materialien aus einfachen Bausteinen
Die Bausteine, aus denen sich natürliche Polysaccharide zusammensetzen, sind in der Regel eine begrenzte Auswahl von Einfachzuckern. Trotzdem weisen natürliche Polysaccharidmaterialien eine breite Spanne von genau kontrollierten Materialeigenschaften auf, die durch hierarchisch geordnete Strukturen der Moleküle entstehen. Diese Strukturen kommen in der Regel durch regulierte Selbstorganisation der Komponenten zustande.
Ein Beispiel sind Strukturen in Pflanzenzellwänden, die sich aus Zellulose und Hemizellulose-Polysacchariden zusammensetzen. Zellulose bildet dabei lange, aber feine kristalline Fasern aus, die in ein weit ungeordneteres Gerüst aus sogenannten Hemizellulose-Polysacchariden eingebettet sind. Obwohl beide aus ähnlichen chemischen Bausteinen bestehen, ist Zellulose nicht wasserlöslich. Die Fasern weisen einen hohen Grad an Festigkeit auf, während die Hemizellulose ein sehr hohes Quellungsvermögen zeigt. Kombiniert man diese beiden Komponenten in einer bestimmten geometrischen Anordnung, können pflanzliche Materialien wie z. B. Reaktionsholz oder unterschiedliche Samenkapseln feuchtigkeitsabhängige gerichtete Kräfte oder Bewegungen ausführen [2].
Das häufige Vorkommen von Polysacchariden in natürlichen Materialien, ihre biologische Verträglichkeit und die vielfältigen biochemischen und biomechanischen Möglichkeiten eröffnen viele potentielle Anwendungen in der Biomaterialforschung. Um die Eigenschaften dieser vielseitigen Moleküle dabei effizient nutzen zu können, ist es wichtig, die Zusammenhänge zwischen molekularen Wechselwirkungen, Struktur und Funktion zu verstehen.
Materialeigenschaften mit dem Computer verstehen
Computersimulationen bieten eine vielversprechende Möglichkeit, Experimente zu ergänzen und ein detailliertes Bild der Moleküle und deren Wechselwirkungen zu gewinnen. Numerische Simulationsansätze reichen dabei von quantenmechanischen Simulationen über atomistische und mesoskopische Modelle bis hin zu statistischen Kontinuumsbeschreibungen. Abbildung 1 zeigt einen Überblick über die relevanten Größenordnungen für Polysaccharide.
 Simulationsbox mit einem Disaccharid und Wassermolekülen mit atomistischer Auflösung; b) die Dichteverteilung von Wassermolekülen um ein Glukosemonomer zeigt Maxima nahe der OH-Gruppen und einen größeren Abstand der Wassermoleküle von der flachen Ringseite; c) Bildung von Wasserstoffbrücken innerhalb eines Moleküls.")
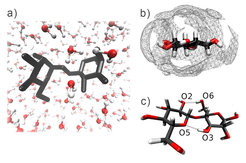
Abb. 2: a) Simulationsbox mit einem Disaccharid und Wassermolekülen mit atomistischer Auflösung; b) die Dichteverteilung von Wassermolekülen um ein Glukosemonomer zeigt Maxima nahe der OH-Gruppen und einen größeren Abstand der Wassermoleküle von der flachen Ringseite; c) Bildung von Wasserstoffbrücken innerhalb eines Moleküls.
Molecular Dynamic (MD) Simulationen, die alle Atome des Systems darstellen (Abb. 2a), ermöglichen ein präzises Verständnis der lokalen Wechselwirkungen einzelner Wasser- und Zuckermoleküle, die für das Quellungsverhalten der Hemi-Zellulose eine wichtige Rolle spielen. Die Wechselwirkungen zwischen den einzelnen Atomen werden in solchen Simulationen als klassische Lennard-Jones, elastische oder elektrische Potenziale modelliert. Dabei ist die genaue Parametrisierung dieser Potenziale, das sogenannte Kraftfeld, von entscheidender Bedeutung. Die meisten Kohlenhydrat-Kraftfelder sind noch verhältnismäßig neu und wurden vor allem im Hinblick auf die Konformationen einzelner Moleküle getestet. Daher ist es zunächst wichtig, die unterschiedlichen Kraftfelder und Wassermodelle auch auf Ihre Zuverlässigkeit bei der Vorhersage von Lösungseigenschaften wie Diffusion oder Aggregationsverhalten zu testen. Hier zeigen sich bei einigen Kraftfeldern erhebliche Qualitätsunterschiede.
Nachdem ein geeignetes Kraftfeld identifiziert ist, können damit Struktur und Dynamik von Wassermolekülen in der unmittelbaren Nähe von typischen Bausteinen der Hemizellulose-Moleküle charakterisiert werden. Ein Beispiel sind Dichteverteilungen der Wassermoleküle um einzelne Zucker, wie in Abb. 2b. Wird ein solches Molekül in Wasser gelöst, kann auch die Änderung der freien Energie berechnet und mit den strukturellen Eigenschaften in Verbindung gesetzt werden. Die Energieunterschiede werden dabei von der Zahl der freien OH-Gruppen dominiert, die langlebige Wasserstoffbrücken mit den Wassermolekülen bilden können. Die Fähigkeit, Wasserstoffbrücken innerhalb des Moleküls zu bilden (Abb. 2c), ist dabei eher nebensächlich. Sie führt aber zu leicht unterschiedlichen Energiewerten für Moleküle mit der gleichen Anzahl von OH-Gruppen [3].
Für die Wasseraufnahme in dichten Polysaccharid-Netzwerken wie z. B. in der Pflanzenzellwand ist das sogenannte chemische Potenzial entscheidender als die Lösungsenergie des Polysaccharids im Wasser. Das chemische Potenzial ist die Energie eines Wassermoleküls im Material relativ zu reinem Wasser. Es steht im direkten Zusammenhang mit dem osmotischen Druck, einer experimentell relativ gut zugänglichen Größe. In Computersimulationen lässt sich der osmotische Druck ganz ähnlich messen wie im Experiment: Die Zuckermoleküle werden durch ein Potenzial in einem Teilbereich der Simulationsbox festgehalten, während der Rest des Systems nur Wasser enthält. Das Potenzial entspricht einer semipermeablen Membran. Die Kräfte auf dieser virtuellen Wand können über die Dauer der Simulation nachverfolgt werden [4]. Über diese Größen kann man dann Rückschlüsse über das Quellverhalten von Systemen mit unterschiedlichen molekularen Strukturen und Anordnungen ziehen.
Coarse-Graining: größere Skalen gegen Genauigkeit
 Freie-Energie-Karten für die Torsionswinkel φ und ψ, die flexibelsten Freiheitsgrade in Disacchariden; b) Für eine Darstellung des Polymers anhand von φ und ψ reduziert sich der Dimer auf eine Folge von drei Bindungen pro Monomer (rot & cyan). Für lange Polymere sollte auch das von den Ringen ausgeschlossene Volumen (graue Kugeln) berücksichtigt werden; c) CG-Abbildung eines Monomers auf drei Wechselwirkungszentren.")
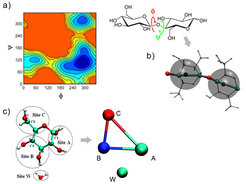
Computermodelle mit atomarer Auflösung geben ein detailliertes Bild der Systeme, können aber nur relativ kleine Systeme und kurze Zeitskalen erreichen. Die Hemizellulose-Polysaccharide bestehen dagegen aus 500-1500 Monomeren, so dass selbst die Simulation einzelner Moleküle mit atomarer Auflösung nicht praktikabel ist. Um den Zusammenhang zwischen der molekularen Struktur und den Quellungseigenschaften der Hemizellulose und den daraus entstehenden Kräften beschreiben zu können, werden reduzierte Modelle mit weniger Freiheitsgraden notwendig. Diese sogenannten „Coarse-Grained (CG)“ Modelle vereinen Gruppen von Atomen oder Materialstücke zu effektiven Wechselwirkungszentren. Die große Herausforderung in solchen Modellen ist eine angemessene Darstellung der Wechselwirkungen zwischen diesen Teilchen, die die zugrundeliegenden physikalischen Prinzipien wiederspiegelt.
Eine Möglichkeit, die Konformationen einzelner Polysaccharide auf großen Längenskalen zu erzeugen, ist, die Beschreibung auf die flexibelsten Freiheitsgrade, die Torsionswinkel der Verbindungen zwischen den verhältnismäßig steifen Ringzuckern, zu reduzieren (Abb. 3). Energielandschaften (Abb. 3a) für die Kombinationen dieser Winkel stammen dabei aus atomistischen Simulationen von kleinen Molekülen. Dabei wird angenommen, dass diese Winkelkarten überwiegend von den direkt benachbarten Monomeren abhängen. Der wichtige Einfluss des Wassers auf die Polymere ist in einem solchen Modell implizit enthalten, da die Winkelkarten im Wasser gemessen werden.
Für dichtere Systeme gewinnen die nicht-kovalenten Wechselwirkungen zwischen Zucker-Monomeren an Bedeutung, so dass diese wieder zum Modell hinzugefügt werden sollten. Dazu kommt, dass für die Modellierung der Wasseraufnahme eine explizite Darstellung der Wassermoleküle vorteilhaft ist. Strategien, um die Wechselwirkungspotenziale für das vereinfachte Model aus den hochaufgelösten Simulationen herzuleiten, sind oft iterative Methoden, die bestimmte Größen, wie Kräfte, Energien oder Strukturen möglichst genau reproduzieren. Die vielversprechendste Möglichkeit, Potenziale zu parametrisieren, ist eine Methode, die die Kräfte im atomistischen System reproduziert [5]. Ein großer Vorteil dieser Strategie ist es, dass sich der osmotische Druck für dieses CG-Modell wie oben über die Kräfte messen und mit experimentellen Daten vergleichen lässt.
Ein wichtiger Punkt für die Parametrisierung dieser Potenziale ist ihre Übertragbarkeit. Um Nutzen aus dem reduzierten Modell ziehen zu können, ist es notwendig Monomerbausteine zu konstruieren, aus denen längere und komplexere Polymere aufgebaut werden können. Diese Übertragbarkeit ist bei systematischen Methoden nicht automatisch gegeben, da die Potenziale die freie Energielandschaft des Systems wiederspiegeln und somit zum Teil stark systemabhängige Größen beinhalten.
Literaturhinweise
2nd Edition, Cold Spring Harbor Laboratory Press, Cold Spring Harbor/N.Y. 2009, 784 p.
Faraday Discussions 139, 275-282 (2008)