Strukturentstehung und -änderung bei Faltung und Funktion von Proteinen
Forschungsbericht (importiert) 2008 - Max-Planck-Institut für Kolloid- und Grenzflächenforschung
Struktur und Funktion von Proteinen
Proteine sind Kettenmoleküle, die aus einzelnen Aminosäuren aufgebaut sind. Die genaue Sequenz der 20 verschiedenartigen Aminosäuren innerhalb der Proteinkette bestimmt dabei, in welche spezielle Struktur sich ein Protein faltet [1] (Abb. 1). Die dreidimensionale Struktur bestimmt wiederum die Funktionen der Proteine, die von der mechanischen Stabilisierung von Zellen und Geweben über die Katalyse von chemischen Reaktionen bis zum Transport von Molekülen reichen. Doch nur korrekt gefaltet kann ein Protein seine Funktion erfüllen. Fehler bei der Faltung können zu Proteinzuständen führen, die schwere Krankheiten wie Alzheimer, Parkinson oder das Creutzfeldt-Jakob-Syndrom hervorrufen.

Der Übergangszustand der Faltung
Viele kleine Proteine falten ohne experimentell nachweisbare metastabile Zwischenzustände. Der erstaunlich direkte Übergang dieser Proteine vom ungefalteten in den gefalteten Zustand wird als „Zwei-Zustands-Faltung“ bezeichnet. Entscheidend für die Faltungsdynamik dieser Proteine ist der Übergangszustand zwischen dem ungefalteten und dem gefalteten Zustand. Da dieser Übergangszustand als instabiler Zustand extrem kurzlebig ist, kann er nicht direkt beobachtet werden. Einen indirekten Zugang ermöglichen jedoch Mutationen eines Proteins, bei denen einzelne Aminosäuren ausgetauscht werden. Die Mutationen verändern geringfügig die Übergangszustandsbarriere und damit die Faltungs- und Entfaltungszeiten des Proteins [2]. Die zentrale Frage ist nun, wie sich der Übergangszustand aus den beobachteten Änderungen der Faltungszeit rekonstruieren lässt.
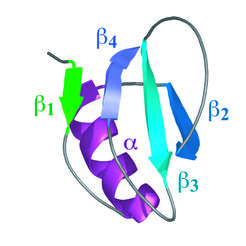
In den letzten Jahren wurde ein neues Modell zur Rekonstruktion von Übergangszuständen aus Mutationsdaten entwickelt [3-5]. In diesem Modell wird der Übergangszustand als ein Ensemble von Übergangsstrukturen beschrieben. Eine zentrale Annahme des Modells ist, dass kooperative Strukturelemente wie α-Helizes und β-Haarnadeln in den einzelnen Übergangsstrukturen entweder vollständig gefaltet oder ungefaltet sind. In dem Modell werden zudem mutationsinduzierte Veränderungen der freien Energie in Komponenten für die Strukturelemente des Proteins aufgeteilt. Die Modellierung der experimentellen Daten verrät, in welchem Grad α-Helizes and β-Haarnadeln im Übergangszustand strukturiert sind. Im Falle der α-Helix des in Abbildung 1 gezeigten Proteins CI2 beispielsweise führt die Modellierung zu dem Ergebnis, dass die Helix im Übergangszustand vollständig ausgebildet, jedoch noch nicht an das β-Faltblatt angelagert ist [4].
Schleifenschließung und Faltungswege
Die überwiegende Mehrheit der Zwei-Zustands-Proteine hat eine Kettenlänge zwischen 60 und 120 Aminosäuren. Doch trotz ähnlicher Länge können sich die Faltungszeiten von
Zwei-Zustands-Proteinen um viele Größenordnungen unterscheiden. Die schnellsten Zwei-Zustands-Proteine falten innerhalb von Mikrosekunden, während die langsamsten mehrere Sekunden zur Faltung benötigen.
Vor einigen Jahren wurde entdeckt, dass sich diese großen Unterschiede in den Faltungszeiten auf Unterschiede in den Proteinstrukturen zurückführen lassen [6]. Schnell faltende Proteine haben Strukturen mit überwiegend lokalen Kontakten zwischen den Aminosäuren. Bei einem lokalen Kontakt zwischen zwei Aminosäuren i und j ist der Sequenzabstand |i-j| der Aminosäuren klein. Im Gegensatz dazu weisen die Strukturen langsam faltender Proteine viele nichtlokale Kontakte mit großem Sequenzabstand auf. Nichtlokale Kontakte können im ungefalteten Zustand nur durch Schließen langer Schleifen in der Proteinkette gebildet werden, während bei lokalen Kontakten nur kurze Schleifen geschlossen werden müssen. Da kurze Schleifen schneller schließen als lange Schleifen, scheinen Schleifenschließungsereignisse für die unterschiedlichen Faltungszeiten verantwortlich zu sein [7].
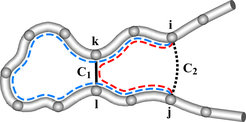
Auch die Faltungswege dieser Proteine lassen sich aus Schleifenschießungsereignissen ableiten [7-9]. Der Faltungsweg eines Proteins ist durch die Reihenfolge bestimmt, in der die Kontakte zwischen den Aminosäuren gebildet werden. Dabei ist zu beachten, dass durch das Schließen von Kontakten Querverbindungen in der Proteinkette entstehen, die die Schleifenlängen bei der Bildung weiterer Kontakte verkürzen (Abb. 2).
. Ohne den Kontakt C1 wäre die Schleife bei der Bildung des Kontakts C2 deutlich länger (blaue Schleife).")
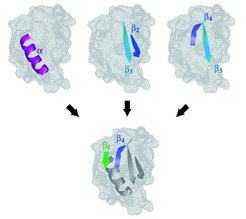
Es zeigt sich, dass Proteine wie ein dreidimensionaler Reißverschluss durch sukzessives Schließen kurzer Schleifen falten können. Auf diesen Faltungswegen bilden sich die Strukturelemente eines Proteins in charakteristischen Reihenfolgen (Abb. 3), aus denen sich häufig auch die grobe Struktur von Übergangszuständen ableiten lassen [7, 9].
 durch sukzessives Schließen kurzer Schleifen. Auf diesem Faltungsweg werden zunächst die α-Helix und die β-Strangverbindungen β2β3 und β3β4 gebildet, in beliebiger Reihenfolge. Diese drei Strukturelemente verkürzen die Schleife, die dann bei der Bildung der Strangverbindung β1β4 zwischen den beiden Proteinenden geschlossen werden muss.")
Strukturänderungen bei der Bindung von Proteinen
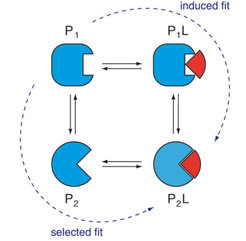
Die biologische Funktion vieler Proteine erfordert kleinere oder größere Strukturänderungen im gefalteten Zustand. Solche Strukturänderungen können beispielsweise beim Binden von Proteinen an andere Proteine oder kleinere Moleküle auftreten. Das belegen experimentell bestimmte Strukturen von Proteinen in gebundenen und ungebundenen Zuständen. Wie die Strukturänderungen genau vonstatten gehen, ist jedoch experimentell nicht direkt beobachtbar. Bereits vor etwa 50 Jahren wurde vorgeschlagen, dass der Bindungspartner eines Proteins, der sogenannte Ligand, durch das Anbinden die Strukturänderung induziert (induced fit). Im Gegensatz dazu wurde vor einigen Jahren postuliert, dass Proteine bereits im ungebundenen Zustand verschiedene Strukturen durchlaufen, und der Ligand lediglich die passende Struktur „wählt“ und durch das Anbinden stabilisiert (selected fit).
Diese beiden Bindungswege lassen sich in einem einfachen Modell mit vier Zuständen veranschaulichen (Abb. 4). In diesem Modell kann das Protein zwei Strukturen P1 und P2 annehmen. Ohne Ligand hat das Protein im Grundzustand die Struktur P1, und in einem angeregten Zustand die Struktur P2. Mit gebundenem Liganden hingegen nimmt das Protein im Grundzustand P2L die Struktur P2 an. Die beiden Grundzustände sind durch zwei verschiedene Bindungswege miteinander verbunden. Auf dem induced-fit-Bindungsweg bindet das Protein den Liganden zunächst im Grundzustand P1. Auf dem selected-fit-Bindungsweg hingegen bindet der Ligand im angeregten Zustand P2. Die Berechnung der Bindungskinetik in diesem Modell zeigt nun, dass nur die Bindungsrate des selected-fit-Weges von der Energiedifferenz zwischen Grundzustand P1 und angeregtem Zustand P2 abhängt [10]. Mit Mutationen des Proteins, die diese Energiedifferenz beeinflussen, sollte sich deshalb unterscheiden lassen, ob ein Protein einen Liganden per induced fit oder per selected fit bindet.